ORIGINAL ARTICLE
JOP. J Pancreas (Online) 2014 Sep 28; 15(5):465-474.
Nicotine Alters the Proteome of Two Human Pancreatic Duct Cell Lines
Joao A Paulo
Department of Cell Biology, Harvard Medical School, Boston, USA
ABSTRACT
Context Cigarette smoking is a known risk factor of pancreatic disease. Nicotine - a major cigarette tobacco component - can traffic through the circulatory system and may induce fibrosis and metastasis, hallmarks of chronic pancreatitis and pancreatic adenocarcinoma, respectively. However, at the biomolecular level, particularly in pancreatic research, the effects of nicotine remain unresolved. Methods The effects of nicotine on the proteomes of two pancreatic duct cell lines–an immortalized normal cell line (HPNE) and a cancer cell line (PanC1)- were investigated using mass spectrometry-based proteomics. For each cell line, the global proteomesof cells exposed to nicotine for 24 hrswere compared with untreated cells in triplicate using 6-plex tandem mass tag-based isobaric labeling techniques. Results Over 5,000 proteins were detectedper cell line. Of these, over 900 proteins were differentially abundant with statistical significance (corrected P-value <0.01) upon nicotine treatment, 57 of which were so in both cell lines. Amyloid precursor protein, previously observed to increase expression in pancreatic stellate cells when exposed to nicotine, was also up-regulated in both cell lines.In general, the two cell lines varied in the classes of proteins altered by nicotine treatment, supporting published evidence that nicotine may play different roles in the initiation and progression of pancreatic disease. Conclusions Understanding the underlying mechanisms associating nicotine with pancreatic function is paramount to intervention aiming to retard, arrest, or ameliorate pancreatic disease.
INTRODUCTION
In the United States, pancreatic cancer is the fourth most common cause of death due to cancer and has an average one and five year survival rate of 25% and 5%, respectively [1]. In pancreas adenocarcinoma - the major form of pancreatic cancer - tumor cells arise from pancreatic ductal epithelium. Pancreatic ductal adenocarcinoma has a mortality rate of nearly 100% within 2 years and comprises over 90% of all pancreatic cancers [2]. Pancreatic duct cells are epithelial cells that line the branched tubes of the organ, secrete bicarbonate, and thus conduct enzymes produced by acinar cells into the duodenum [3]. Delineating the molecular mechanisms regulating pancreatic duct cell function, in addition to alterations produced by cellular stresses, such as nicotine, will add to our general understanding of the disease.
One of the most prominent risk factors correlated with the development of pancreatic cancer is cigarette smoking [4, 5]. However, only limited research has been performed to investigate this association. Cigarette smoking may promote the development of chronic pancreatitis by increasing disease severity and pancreatic calcifications which can further damage the pancreas [6]. Elucidating the underlying cellular events promoted by smoking in the development of pancreatic disease is vital to timely intervention aimed at counteracting the disease.
Many of the toxic compounds found in tobacco smoke are absorbed into the blood stream and several have been detected in pancreatic fluid [7]. Cigarette smoke is an admixture of over 4,000 compounds, for which over 50 are suspected carcinogens [8]. Although not directly implicated in cancer, nicotine is a major toxic component of tobacco. In addition, cigarette smoking cessation programs often rely on nicotine replacement therapy (NRT), such as transdermal patches, nasal sprays or emerging e-Cigarette technology. Nicotine is readily absorbed by the lungs as well as other, more distal organs via systemic circulation [9]. Nicotine interacts with a population of nicotinic acetylcholine receptors (nAChR), with varying affinities and downstream effectors [10] and as such is as a contributing factor to myocardial [11, 12] , atrial [13], pulmonary [14, 15] and biliary fibrosis [16] diseases. The binding of nicotine to cell surface receptors transduces extracellular signals to the intracellular space resulting in ion transport and/or initiation of phosphorylation cascades and other signaling pathways [17-19].
Various cancers and chronic diseases have been linked to nicotine, yet the mechanisms of such are poorly understood [20, 21]. Concerning pancreatic disease, cigarette smoking may induce fibrosis and metastasis, hallmarks of chronic pancreatitis and pancreatic adenocarcinoma, respectively [22] . Evidence suggests that protein expression is altered in pancreatic duct cells (PaDC), and such cellular changes are implicated in pancreatic cancer and chronic pancreatitis [23]. However, to date, no comprehensive analysis of the effects of nicotine on PaDC in an in vitro environment has been published. In various cell types, nicotine is known to induce alterations in protein expression that affect cellular proliferation, chemotaxis, and attachment to various surfaces [24, 25].
Here, I focus on changes in the global proteome of cultured pancreatic cells when subjected to nicotine treatment. I investigate two PaDC cell lines, first a normal epithelial cell line, HPNE (human pancreatic Nestin-expressing), and second a well-studied pancreatic cancer cell line, PanC1 (pancreatic cancer 1). Identifying differences in proteins that are altered in abundance under nicotine stress is an initial step to gain a comprehensive understanding of the cellular physiology regulating the development and progression of pancreatic disease. Research such as that presented herein may broaden our knowledge of the effects of nicotine on the pancreas and further validation via targeted studies may provide targets for drug therapies aiming to retard or reverse the clinical manifestations of pancreatic disease.
MATERIALS AND METHODS
Materials
Dulbecco's modified Eagle's-F12 medium (DMEM/F12; 11330) was purchased from Gibco (Carlsbad, CA). Fetal bovine serum (FBS; F0392) was purchased from Sigma (St. Louis, MO). CellStripper (25-056-CL) for non-enzymatic cell dislodgement was purchased from Mediatech (Manassas, VA). Tandem mass tag (TMT) isobaric reagents were from Thermo Scientific (Rockford, IL). Water and organic solvents were purchased from J.T. Baker (Center Valley, PA). (-)-Nicotine (≥99%) (N3876) was purchased from Sigma (St. Louis, MO). Sequencing-grade modified trypsin (V5111) was obtained from Promega (Madison, WI). Unless otherwise noted, other reagents and solvents were from Sigma-Aldrich and Burdick & Jackson, respectively. Primary antibody against amyloid precursor protein (ab15272) was from Abcam (Cambridge, MA), while secondary horseradish peroxidase anti-rabbit antibody (sc-2313) was from SantaCruz Biotechnology (Santa Cruz, CA).
Cell Lines
The PaDC cell line, hTERT-HPNE (CRL-4023), was purchased from ATCC (Manassas, VA). These cells were immortalized by transduction with the catalytic subunit of human telomerase (hTERT) [26]. The well-established tumor-cell line from a human carcinoma of the exocrine pancreas, PanC1, was also purchased from ATCC [27]. Both cell lines are adherent epithelial pancreatic duct cells from males. However, these cells are not isogenic as they have unique origins and have been isolated differently. Cells lines were passaged 5 times in our hands.
Experimental Strategy
The experimental strategy was outlined in Figure 1. This procedure was performed in parallel for both HPNE and PanC1 cell lines. Cells were lysed and proteins were extracted via methanol-chloroform precipitation and then digested with LysC and trypsin. Each sample was labeled with a specific TMT isobaric tag. The pooled sample was fractionated by basic pH reversed-phase (BpRP) chromatography and subjected to LC-MS3 analysis.
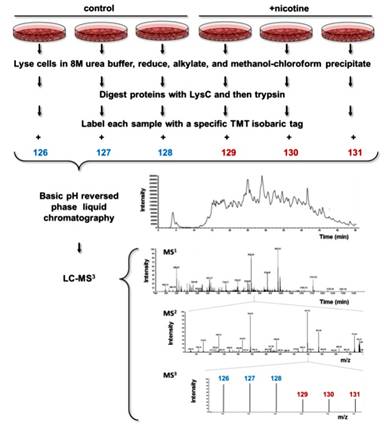
|
Figure 1.Workflow of the quantitative mass spectrometry-based TMT MS3 experiments.Following cell lysis and protein extraction, proteins were digested with LysC and then trypsin. Each sample was labeled with a specific TMT isobaric tag (control cells were labeled with 126-128 colored in blue and PanC1 with 129-131 colored in red) and then combined. The pooled sample was fractionated by basic pH reversed-phase (BpRP) liquid chromatography and subjected to LC-MS3 analysis. This procedure was performed in parallel for both HPNE and PanC1 cell lines. |
Cell Growth and Harvesting of Pancreatic Duct Cells
Methods of cell growth and propagation followed previously utilized techniques [28, 29]. In brief, cells were propagated in Dulbecco's modified Eagle's-F12 medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Upon achieving 85-90% confluency, the growth media was aspirated and the cells were washed 3 times with ice-cold phosphate-buffered saline (PBS). This cell density was chosen so as to maximize the amount of cells for harvesting while ensuring that the cells are proliferating. Assigned cell culture dishes were supplemented with 1 µM nicotine and control cell culture dishes were mock treated with an equal volume of sterile deionized water. Twenty-four hours after the addition of fresh media, the cells were dislodged with a non-enzymatic reagent, harvested by trituration following the addition of 10 mL PBS, pelleted by centrifugation at 3,000 x g for 5 min at 4°C, and the supernatant was removed. One milliliter of buffer containing TBSp (50 mM Tris, 150 mM NaCl, pH 7.4 supplemented with 1X Roche Complete protease inhibitors), 1% Triton X-100 and 0.5% SDS was added to each 10 cm cell culture dish.
Cell Lysis and Protein Digestion
Cells were homogenized by 12 passes through a 27 gauge (1.25 inches long) needle and incubated at 4°C with gentle agitation for 1 hr. The homogenate was sedimented by ultracentrifugation at 100,000 x g for 60 min at 4°C. Protein concentrations were determined using the bicinchoninic acid (BCA) assay (ThermoFisher Scientific). Proteins were subjected to disulfide bond reduction with 5 mM dithiotreitol (37°C, 25 min) and alkylation with 10 mM iodoacetamide (room temperature, 30 min in the dark). Excess iodoacetamide was quenched with 15 mM dithiotreitol (room temperature, 15 min in the dark).
Methanol-chloroform precipitation was performed prior to protease digestion. In brief, 4 parts of neat methanol was added to each sample and vortexed, 1 part chloroform was added to the sample and vortexed, and 3 parts water was added to the sample and vortexed. The sample was centrifuged at 10,000 RPM for 15 min at room temperature and subsequently washed twice with 100% acetone, prior to air-drying.
Samples were resuspended in 1 M urea, 50 mM HEPES, pH 8.5. Protein concentrations were determined using the BCA assay. Approximately 150μg of protein was digested at 37°C for 3 hrs with LysC protease at a 100:1 protein-to-protease ratio. Trypsin was then added at a 100:1 protein-to-protease ratio and incubated overnight for 37°C. This reaction was then quenched with 1% formic acid, subjected to C18 solid-phase extraction (SPE) (Sep-Pak, Waters) and subsequently vacuum-centrifuged to near-dryness.
Tandem Mass Tag Labeling
In preparation for TMT labeling, desalted peptides were dissolved in 200 mM HEPES, pH 8.5. Peptide concentrations were determined using the microBCA assay. Approximately 100 μg of peptides from each sample were labeled with TMT reagent.
TMT reagents were dissolved in anhydrous acetonitrile and added to the peptides to achieve a final acetonitrile concentration of 30% (v/v). Following incubation at room temperature for 1 hr, the reaction was quenched with hydroxylamine at a final concentration of 0.3% (v/v). The TMT-labeled samples were combined at a 1:1:1:1:1:1 ratio. The samples were vacuum centrifuged to near dryness and subjected to C18 solid-phase extraction (Sep-Pak, Waters).
Offline Basic pH Reversed-phase (BpRP) Fractionation
The peptide mixture was separated into a total of 96 fractions using an Agilent 1100 quaternary pump equipped with a degasser and a photodiode array (PDA) detector (set at 220 and 280-nm wavelength) from ThermoFisher (Waltham, MA). A 50 min linear gradient from 5% to 35% acetonitrile in 10mM ammonium bicarbonate pH8 at a flow rate of 0.8 mL/min separated the peptides on an Agilent 300Extend C18 column (5 μm particles, 4.6 mm ID and 220 mm in length). The 96 fractions were consolidated into 12 using a checkerboard pattern. Samples were subsequently acidified with 1% formic acid and vacuum centrifuged to near dryness. Each fraction was desalted via StageTip, dried with vacuum centrifugation, and reconstituted in 5% acetonitrile, 5% formic acid for LC-MS/MS processing.
Liquid Chromatography and Tandem Mass Spectrometry
The mass spectrometry data were collected using an Orbitrap Velos Pro mass spectrometer (Thermo Fisher Scientific, San Jose, CA) coupled to an Accella 600 liquid chromatography (LC) pump (Thermo Fisher Scientific) and a Famos autosampler (LC packings). Peptides were separated on a 100 μm inner diameter microcapillary column packed with ∼0.5 cm of Magic C4 resin (5 μm, 100 Å, Michrom Bioresources) followed by ∼20 cm of Maccel C18 resin (3 μm, 200 Å, Nest Group). For each analysis, ~1 μg of peptide was loaded onto the column.
Peptides were separated using a 3 hr gradient of 6 to 30% acetonitrile in 0.125% formic acid with a flow rate of ∼300 nL/min, using an MS3-based TMT method [30]. The scan sequence began with an MS1 spectrum (Orbitrap analysis, resolution 60,000, 300−1500 Th, automatic gain control (AGC) target 1 × 106, maximum injection time 150 ms). The top ten precursors were then selected for MS2/MS3 analysis. MS2 analysis consisted of collision-induced dissociation (CID), quadrupole ion trap analysis, automatic gain control (AGC) 2 × 103, NCE (normalized collision energy) 35, q-value 0.25, and maximum injection time 100 ms. Following acquisition of each MS2 spectrum, an MS3 spectrum was collected with a recently described method in which multiple MS2 fragment ions were captured in the MS3 precursor population using isolation waveforms [30] with multiple frequency notches. MS3 precursors were fragmented by HCD and analyzed using the Orbitrap (NCE 50, Max AGC 1.5 × 105, maximum injection time 250 ms, isolation specificity 0.8 Th, resolution was 30,000 at 400 Th).
Data Analysis
Mass spectra were processed using a Sequest-based in-house software pipeline. First, mass spectra were converted to mzXML using a modified version of ReAdW.exe. Database searching included all entries from the human IPI database (v. 3.87), which was concatenated with a database composed of all protein sequences in reversed order. Searches were performed using a 50 ppm precursor ion tolerance. Product ion tolerance was set to 0.03 Th. TMT tags on lysine residues and peptide N termini (+229.163 Da) and carbamidomethylation of cysteine residues (+57.021 Da) were set as static modifications, while oxidation of methionine residues (+15.995 Da) was set as a variable modification.
Peptide-spectrum matches (PSMs) were altered to a 1% false discovery rate (FDR) [31]. PSM filtering was performed using a linear discriminant analysis, as described previously [32], while considering the following parameters: XCorr, ΔCn, missed cleavages, peptide length, charge state, and precursor mass accuracy. The signal-to-noise (S/N) ratio for each TMT channel was extracted for TMT-based reporter ion quantitation, and the closest matching centroid to the expected mass of the TMT reporter ion was determined.
The search space for each reporter ion was limited to a range of 0.002 Th to prevent overlap between the isobaric reporter ions. For protein-level comparisons, PSMs were identified, quantified, and then collapsed further to a final protein-level FDR of 1%. Furthermore, protein assembly was guided by principles of parsimony to produce the smallest set of proteins necessary to account for all observed peptides. Proteins were quantified by summing reporter ion counts across all matching PSMs. PSMs with poor quality MS3 spectra (more than 4 TMT channels missing and/or fewer than 75 total reporter ions) or no MS3 spectra were excluded from quantification.
Student t-tests were then used to identify proteins that were differentially expressed and the method of Benjamini and Hochberg was subsequently applied to control for multiple testing [33]. A Benjamini-Hochberg-corrected P-value < 0.01 was considered statistically significant, and a secondary threshold requiring a ±1.5 fold change was used. Protein quantitation values were exported for further analysis in Excel or Mathematica. Hierarchical clustering was performed using Multi Experiment Viewer [34].
Data Access
RAW files will be made available upon request.
RESULTS
Over 5,000 Proteins Were Detected per Cell Line Using TMT-MS3 Analysis
Following the schema as outlined in Figure 1, I detected 5211 proteins in the HPNE cell line and 5930 in the PanC1 cell line. Of these, 4243 proteins were present in both cell lines, while 968 were unique to HPNE and 1687 were unique to PanC1 (Figure 2a). The significance threshold was set at a Benjamini-Hochberg corrected P-value of <0.01 and only those proteins with a fold change of ±1.5 were considered. Using these criteria, 315 proteins were significantly different in abundance upon nicotine treatment within the HPNE cell line (221 up-regulated and 94 down-regulated). Similarly, 731 proteins were significantly different within the PanC1 cell line (239 up-regulated and 492 down-regulated), as illustrated in Table 1. Comparing both cell lines, only 57 of the 989 non-redundant proteins with statistically significant alterations in abundance in at least one cell line were significantly altered in both cell lines (Figure 2b).
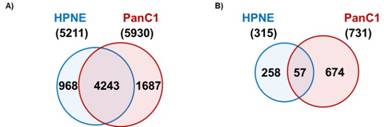
|
Figure 2.Protein overlap between cell lines. A)Venn diagram illustrating that a total of 5211 proteins were detected in the HPNE cell line, while 5930 proteins were detected in the PanC1 cell line, of which there was an overlap of 4243 proteins. B) Venn diagram representing the number of proteins demonstrating statistically significant differences (P<0.01 and fold change ±1.5) in abundance upon nicotine stress. |
|
Table 1. Summary of proteins quantified and those differentially abundant within the HPNE and PanC1 cell lines. |
|||
|
Number of proteins detected |
Statistically significant |
||
|
up |
down |
||
|
HPNE |
5,211 |
221 |
94 |
|
PanC1 |
5,930 |
239 |
492 |
Nicotine Altered Protein Abundance Levels in Both HPNE and PanC1 Cell Lines
I have illustrated protein fold changes and statistical significance graphically as volcano plots for HPNE (Figure 3a) and PanC1 (Figure 3b). In these plots, the -log10 of the Benjamini-Hochberg corrected P-value was plotted against the log2 ratio of the protein abundance. This ratio was calculated using the average for the three nicotine-treated replicates of the summed peptide signal to noise for each protein divided by the corresponding average of the control replicates. In each volcano plot, the upper leftmost section represented those proteins with a 1.5-fold or greater decrease in abundance and a significant P-value, while the upper rightmost section represented those proteins with a 1.5-fold or greater increase in abundance, again with a significant P-value.
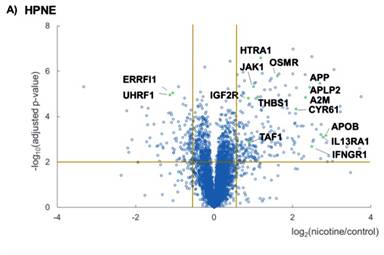
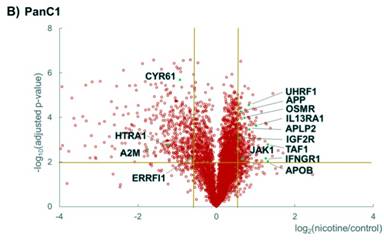
In efforts to discover pathways that were modulated upon nicotine treatment, I then performed Panther pathway analysis via DAVID (Database for Annotation, Visualization and Integrated Discovery) [35]. Here, I highlighted pathways that were involved in JAK/STAT signaling, nAChR-binding, cytokine receptor binding and cell proliferation. I chose to limit the discussion to a set of pathways that were relevant to nicotine–induced signaling (JAK/STAT signaling and nAChR-binding) alterations as well as pancreatic disease (cytokine receptor binding and cell proliferation). Although I have associated these proteins to specific pathways, most proteins partake in more than one pathway, either with a minor or major role. For example, I have classified, E3 ubiquitin-protein ligase (UHRF1) as involved in cell proliferation, but it also has a role in chromatin structure and gene expression and is an oncogene for hepatocellular carcinoma [36]. Therefore, the pathway categories listed are not exclusive for that particular set of proteins.
Protein abundance data for the four pathways that I have chosen to investigate were illustrated in Supplemental Figure 1. First, proteins involved with JAK/STAT signaling include Interferon gamma (IFNGR1), Interleukin-13 receptor subunit alpha-1 (IL13RA1), Tyrosine-protein kinase (JAK1), and Oncostatin-M-specific receptor subunit beta (OSMR) (Supplemental Figure 1a). Second, proteins demonstrating cytokine receptor binding include Cation-independent mannose-6-phosphate receptor (IGF2R), Alpha-2-macroglobulin (A2M), Cysteine rich protein 61 (CYR61), and Thrombospondin-1 (THBS1) (Supplemental Figure 1b). Third, proteins involved in cell proliferation include High-temperature requirement A serine peptidase (HTRA1), E3 ubiquitin-protein ligase (UHRF1), ERBB receptor feedback inhibitor 1 (ERRFI1), and Transcription initiation factor 1 (TAF1) (Supplemental Figure 1c). Fourth, suspected nAChR binding partners include Amyloid-like protein 2 (APLP2), Apolipoprotein B (APOB), and Amyloid beta A4 protein (APP) (Supplemental Figure 1d). Some functional overlap was present within these groups, as CYR61 and HTRA1 can have roles in both cell proliferation and cytokine receptor binding, while OSMR also functions in cytokine receptor binding, as well as JAK/STAT signaling.
Although the abundance of most proteins above were altered either up or down in both cell lines, a few proteins changed in opposing directions. In both cell lines, all three of the suspected nAChR binding proteins were of higher abundance in the nicotine-treated samples than the untreated samples. In contrast, A2M, CYR61, HTRA1, and THBS1, all of which have functions in cytokine receptor binding, were of higher abundance in nicotine-treated HPNE cells compared to control, however the opposite was true for PanC1. Conversely, UHRF1 was of relatively lower abundance in HPNE cells than PanC1 upon nicotine treatment. The mechanistic relationship between nicotine and the alteration in proteins expression requires further clarification. Investigating signaling cascades associated with these proteins may explain such variations.
Nicotine Affected the Abundance of Different Sets of Proteins in the Two Cell Lines
In total, 989 proteins showed statistically significant differences in abundance in at least one cell line upon treatment with nicotine. Hierarchical clustering was performed subsequently on these proteins. Data for each protein across quantified channels for the HPNE cell line (Figure 4a) were normalized with respect to the average wildtype abundance levels, as were data for PanC1 (Figure 4b) independently. In both cell lines, samples clustered first by triplicate, that is, untreated cells clustered together, as did the nicotine-treated cells. I divided the clustering diagrams into 5 sections according to the direction of changes in protein abundance. The DAVID bioinformatics resource allows data mining with a comprehensive set of biologically-relevant functional annotation tools [35]. Using this tool, I assigned the major enriched protein class categories for each cluster, which are listed to the right of the heat maps in Figure 4.
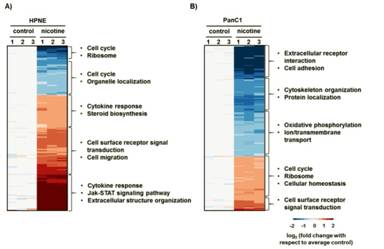
|
Figure 4. Statistically significant alterations in pancreatic duct cell proteins. Heat map of the A) 315 proteins that were statistically significant in the HPNE cell line and B) 731 proteins that were statistically significant in the PanC1 cell line. Enriched gene ontology classifications are listed to the right of each heat map. The heat maps compare the protein abundance ratios of all samples with respect to that of the average control cells. |
Only 57 of the 989 proteins with statistically significant alterations in abundance in at least one cell line were significantly altered in both cell lines (Figure 3b). I then divided the proteins into four groups corresponding to the direction of protein expression in terms of nicotine treatment to controls for both cell lines (Figure 4), that is, up in both, down in both, down in HPNE but up in PanC1, and up in HPNE but down in PanC1. Of these proteins, 28 showed altered expression in the same direction. More specifically, 18 were of higher abundance upon treatment with nicotine, and 10 of lower abundance. Conversely, 29 proteins were significantly altered in opposite directions. Of these, seven were of lower abundance in HPNE and higher abundance in PanC1, while 22 were of higher abundance in HPNE and lower abundance in PanC1 relative to nicotine treatment. The differences in abundance changes provide support for cell line-specific effects of nicotine.
DISCUSSION
The data indicate that nicotine extensively alters protein abundance in pancreatic duct cells with distinctive proteins being differentially regulated in HPNE and PanC1 cell lines. An isobaric tandem mass tag mass spectrometry approach detected 5211 proteins in the HPNE cell line and 5930 proteins in the PanC1 cell line. Considering both cell lines, I determined 989 proteins to be differentially abundant with statistical significance (Benjamini-Hochberg corrected P-value <0.01 and fold change greater than ±1.5), 57 of which were altered upon nicotine treatment in both cell lines. Nicotine exposure affected the expression of different classes of proteins in both cell lines. Such data may support published evidence that the effects of nicotine may be different in the initiation and progression of the disease [37, 38].
Functional annotations were culled from the DAVID bioinformatics resource to examine the pathways in which the proteins detected may have a role. As proteins may have several different functions in a cell, the categories listed are representative of the whole cluster and may not reflect the major functional category of certain proteins. Of the pathways listed, cell surface receptor signal transduction proteins were up-regulated following nicotine treatment in both cell lines, which is consistent with the frequently observed effect in various systems [39, 40]. In PanC1, certain membrane proteins appear to be down-regulated upon nicotine treatment, such as those involved in cell adhesion and transmembrane/ion transport as well as oxidative phosphorylation [41]. In HPNE, up-regulated proteins included those involved in cytokine response, steroid biosynthesis, cell migration, extracellular structure organization and the JAK/STAT signaling pathway, all of which have been implicated as cellular responses to nicotine [17, 20, 42]. Cell division, ribosomes, and certain cell cycle proteins trended down in HPNE and up in PanC1, emphasizing the different effects that nicotine has on the cell lines. Overall, these data show that altered proteins spanned a wide-range of cellular functions, albeit differently between cell types. Such differences were expected as PanC1 cells are substantially differentiated from non-cancerous PaDC (HPNE) both functionally and morphologically.
Among the functional annotations highlighted by David, cytokine response is of particular interest in relation to its cellular interactions with the surrounding microenvironment. Cytokines are known to affect the development and progression of pancreatic cancer and chronic pancreatitis [43, 44]. Studies using pancreatic stellate cells, for example, have underscored the expression of several growth factors, chemokines and cytokines, known to participate in inflammatory and fibrotic responses to pancreatic injury. These responses are often precursors to malignant and pre-malignant lesions [43, 45, 46]. Moreover, nicotine has also been implicated in the release and expression modulation of cytokines in a variety of cellular systems [47, 48]. In this study, several differentially expressed proteins were involved in cytokine receptor binding and JAK/STAT signaling (Supplementary Figure 1a). The expressed regulatory factors controlling the cellular functions of pancreatic cells represent potential diagnostic and therapeutic targets. However, the effects of nicotine on the expression and cellular regulation of cytokines and associated pathways have not been thoroughly investigated and merit further study.
Related to the inflammatory and fibrotic responses, the amyloid precursor protein (APP) was up-regulated in both cell lines treated with nicotine (Supplementary Figure 1d). Western blotting against APP, using actin as a protein loading control (Supplemental Figure 1e), validated the mass spectrometric data which showed similarly increased levels of APP expression in both cell types when treated with nicotine. APP is well studied in the nervous system and is linked to the development of Alzheimer’s disease [49, 50]. APP has been identified previously in an interactome study of the α7 nAChR from mouse brain tissue [19] and has been shown in several studies to bind and modulate the activity of this receptor [51, 52]. In addition to expression in the nervous system, APP has been identified at high levels in the pancreas, epidermis, and thyroid [53, 54]. In neurons, APP is suspected to have an effect in cell growth and process extension [49], although the physiological role of APP in the pancreas and other organs has not been well defined. However, evidence has suggested APP may be involved in inflammatory and cytokine-mediated response [55], which correlates well with the effects of nicotine on cytokine-related proteins as observed herein. APP has been noted previously as being up-regulated in nicotine-treated pancreatic cells in a cross-species analysis, which included pancreatic stellate cells from mouse, rat, and human species [56]. Moreover, an in vitro study using pancreatic ductal epithelial cells has indicated that increased APP expression in pancreatic cancer may influence cellular proliferation [57]. This study revealed the expression and cellular localization of APP and associated proteins in pancreatic ductal epithelial cell lines under standard growth conditions without exogenous perturbations (e.g., nicotine treatment). As such, further investigation may lead to a link among nicotine, APP expression, inflammatory response, and pancreatic cancer (Figure 5).
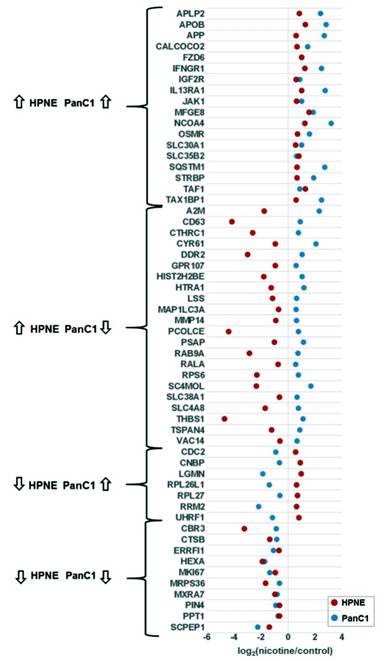
|
Figure 5. Dot plot of proteins illustrating those with statistically significant differences in abundance in both cell lines. Fifty-seven proteins demonstrated statistically significant changes in both cell lines. |
Physiologically, nicotine binds specifically to nicotinic acetylcholine receptors (nAChR) that participate in signal transduction resulting in intracellular phosphorylation cascades. nAChRs have key roles in the chemical transmission of electrical signals at nerve cell synapses and at neuromuscular junctions [58]. These receptors, particularly the α7 subtype, have been found also in non-neuronal cells and are thought to be important regulators of cellular function [59-61]. Nicotine induces cell proliferation in breast, lung, and pancreatic cancer cells. In one study, nicotine increases the levels of vimentin [38]. Another study has demonstrated that nicotine acts via the α3 nAChR and increases the expression of fibrosis-related proteins [62]. Also, in lung cancer cells, nicotine has been shown to activate proliferation by the phosphorylation of Bcl-2 [63]. Ca2+ - induced kinase-based signaling is of particular interest due to the ionotropic nature of nAChRs [64, 65]. More recently, nicotine has been shown to affect secretion in pancreatic acinar cells via calcium signaling [64]. Herein, I have identified proteins differentially expressed upon nicotine treatment that are involved in JAK/STAT signaling and have not been identified previously in the context of PaDC function. Understanding the dynamics of kinase activity and phosphorylation patterns associated with nicotine and PaDC function may provide targets for the early detection of pancreatic disease.
The research outlined herein may be expanded in several diverse directions using the presented techniques. As proteins involved in cytokine receptor binding showed significant changes in protein abundance, we could interrogate the spent culture media for secreted proteins in future analyses. Secretion of cytokines in media, for example, would indicate an autocrine signaling function as has been described for the activation of pancreatic stellate cells [66]. In addition, like nicotine, cellular stress resulting from treatment with metabolic products of nicotine and carcinogens present in cigarette smoke, such as cadmium, toluene, NNK, formaldehyde, may have significant effect on cellular protein expression. Such effects may be similar, more subtle, or more intense than that of nicotine and merit study in future investigations. Moreover, although pancreatic enzymes are conducted to the duodenum via pancreatic duct cells, the acinar cells produce the pancreatic enzymes involved in protein digestion. Dysregulation of these enzymes, particularly characterized by intracellular activation, often results in pancreatitis and later pancreatic cancer [67]. As such, these cells may offer an alternative target for studying the effect of nicotine on the pancreas, which may have a different set of altered pathways.
With the data acquired herein, we can identity proteins that change in abundance, but we cannot assertively determine the molecular mechanisms regulating associated pathway alterations. To understand better these phenomena, posttranslational signaling studies may be informative. Future investigations may include phosphoproteomic analysis of these cell types to study changes resulting from treatment with nicotine. Proteins do not function in isolation, but in complex networks of cross-talking pathways. Phosphorylation events, resulting from the activity of kinases, are major initiators and propagators of signaling events. Evidence supports nicotine as a factor that can alter protein phosphorylation events. For example, nicotine has been shown to induce cell proliferation using a Src and Akt-dependent pathway in pancreatic cancer cells [68] and has a role in the phosphorylation of CREB, ERK, Src, and AKT. Moreover, Shin et al. has shown that nicotine promotes growth of gastric cancers via PKC and ERK1/2 phosphorylation [69]. Such activity resulting from nicotine exposure has not been studied extensively in pancreatic cells and doing so will significantly enhance our understanding of the signaling pathways implicated in the biomolecular alterations triggered by nicotine. The techniques outlined here could be adapted to phosphorylation analysis to allow further interrogation of signaling pathways regulated by nicotine treatment.
Alterations in signaling pathways, may also be heavily dependent on the concentration of nicotine added to the media. For this study, the nicotine concentration was 1 μM, which has been used routinely in gastrointestinal cancer research [70] and for multiple cell lines [38]. This concentration approximates the supposed upper limit in the blood of cigarette smokers, which can vary from 25 to 444 nM [71]. A recent study investigated nicotine-induced cell proliferation in variety of cell lines with the maximum effect of nicotine being observed at a 1 μM concentration [72]. However, protein changes have also been noted at a range of concentrations, as a prior study investigating hepatic stellate cells showed similar alterations at nicotine concentrations ranging from 100 pM to 10 μM [73]. Future studies may explore the consequences of nicotine treatment on PaDC resulting from dose-dependent effects, particularly lower, and physiological, concentrations to show the minimum needed for a significant alteration in abundance, as well as time-dependent protein abundance alterations elicited by nicotine treatment. Tracking the cellular proteomic response to these conditions may provide additional evidence for changes observed in the present study and result in further insights into the mechanisms of disease.
The investigation of signaling pathways can also be enhanced by technological improvements and methodological innovations for mass spectrometric analyses. As proteomic techniques develop further and instrument sensitivity increases, such improvements may refine further the initial investigation described herein. Until recently, only limited targeted mass spectrometry-based strategies were available. These methods require specialized triple quadrupole instruments and intense assay development (multiple and single reaction monitoring, abbreviated MRM and SRM, respectively) [74]. Emerging methods in mass spectrometry may facilitate such strategies and allow for multiplexed, targeted absolute quantitation [75]. Such assays will allow for multiple targets to be analyzed quantitatively, and as such facilitate the simultaneous analysis of several proteins within a pathway. These advancements indicate a positive outlook for future mass spectrometry-based pathway studies.
In conclusion, the data show significant changes in the protein profiles of PaDC following nicotine treatment. Future studies exploring further the impact of nicotine on cellular functions considering dose and time-dependent effects may enable the tracing of alterations in the abundance of specific proteins or modulation of certain pathways thereby expanding our knowledge concerning the effects of nicotine on the pancreas. Moreover, although difficult to obtain and culture, the use of isogenic primary cells that originate from diseased and healthy areas of the same patient would support further the data presented herein. In summary, the data demonstrate that exposure of pancreatic cells to nicotine alters the abundance of certain proteins, the mediation of which may be linked to pancreatic diseases, including pancreatic adenocarcinoma and chronic pancreatitis.
SUPPLEMENTS
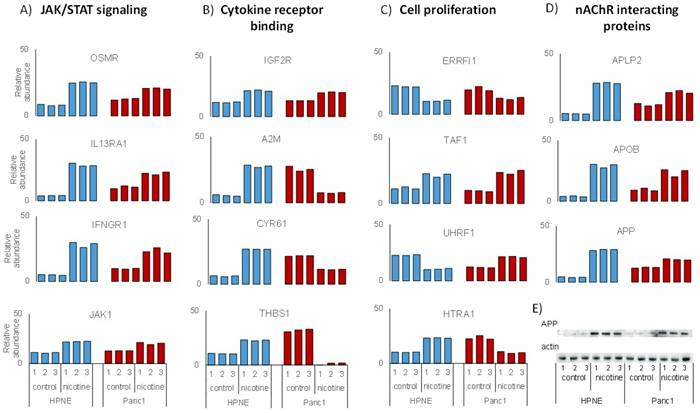
|
Supplemental Figure 1. Examples of statistically significant, differentially abundant proteins following nicotine treatment. The proteins above demonstrate significant changes in abundance in both pancreatic cell lines. These proteins were classified as either A) JAK/STAT signaling proteins, B) nAChR interacting proteins, C) cytokine binding proteins, or D) cell proliferation-related proteins. E) Western blotting analysis probing for amyloid precursor protein (APP,) which is up-regulated in both cell lines upon nicotine treatment, and corresponding actin control blot. |
|
Supplemental Table 1. Proteins quantified in HPNE and PanC1 TMT6-plex analysis. Columns include: Protein identification number, gene symbol, protein description/name, number of peptides identified per protein (peptides), the normalized summed signal to noise for each of the 6 channels (126_sn_norm to 131_sn_norm). |
|
Download: .XLSX format; .PDF format |
Received May 31st, 2014 – Accepted July 10th, 2014
Key words Amyloid; Nicotine; Pancreatic Ducts; Pancreatic Neoplasms; Receptors, Nicotinic
Abbreviations APP: amyloid precursor protein; FBS: fetal bovine serum; PaDC: pancreatic duct cells; TMT: tandem mass tag
Acknowledgements Funds were provided by the following NIH grants: 1 K01 DK098285 (JP) and a grant from the American College of Gastroenterology: ACG – 042103580 (JP). I would also like to thank members of the Gygi Lab at Harvard Medical School for their technical assistance and critical reading of the manuscript, in particular Dr. Steven P Gygi for his mentorship, advice, and use of his mass spectrometers and Robert A Everley for assistance with data collection
Conflict of Interest Authors declare to have no conflict of interest
Correspondence
Joao A Paulo
Department
of Cell Biology
Harvard Medical School
Boston, MA 02115
USA
Phone:
+1-617.432.6767
Fax: +1-617.264.5277
E-mail: joao_paulo@hms.harvard.edu
References
1. Hariharan D, Saied A, Kocher HM. Analysis of mortality rates for pancreatic cancer across the world. HPB (Oxford). 2008; 10: 58-62. [PMID: 18695761].
2. Fuchs CS, Colditz GA, Stampfer MJ, Giovannucci EL, Hunter DJ, Rimm EB, Willett WC, Speizer FE. A prospective study of cigarette smoking and the risk of pancreatic cancer. Arch Intern Med. 1996; 156: 2255-2260. [PMID: 8885826].
3. Grapin-Botton A. Ductal cells of the pancreas. Int J Biochem Cell Biol. 2005; 37: 504-510. [PMID: 15618005].
4. Yadav D, Hawes RH, Brand RE, Anderson MA, Money ME, Banks PA, et al. Alcohol consumption, cigarette smoking, and the risk of recurrent acute and chronic pancreatitis. Arch Intern Med. 2009; 169: 1035-1045. [PMID: 19506173].
5. Law R, Parsi M, Lopez R, Zuccaro G, Stevens T. Cigarette smoking is independently associated with chronic pancreatitis. Pancreatology. 2010; 10: 54-59. [PMID: 20332662].
6. Lin Y, Tamakoshi A, Hayakawa T, Ogawa M, Ohno Y. Cigarette smoking as a risk factor for chronic pancreatitis: a case-control study in Japan. Research Committee on Intractable Pancreatic Diseases. Pancreas. 2000; 21: 109-114. [PMID: 10975702].
7. Prokopczyk, B, Hoffmann D, Bologna M, Cunningham AJ, Trushin N, Akerkar S, Boyiri T. et al. Identification of tobacco-derived compounds in human pancreatic juice. Chem Res Toxicol. 2002; 15: 677-85. [PMID: 12018989].
8. National Toxicology Program. Tobacco-related exposures: tobacco smoking. Report on carcinogens: carcinogen profiles. 2011; 12: 408-410.
9. Chowdhury P, Rayford PL. Smoking and pancreatic disorders. Eur J Gastroenterol Hepatol. 2000; 12: 869-877.[PMID: 10958214].
10. Changeux JP. Nicotine addiction and nicotinic receptors: lessons from genetically modified mice. Nat Rev Neurosci. 2010; 11: 389-401.[PMID: 20485364].
11. Hu N, Guo R, Han X, Zhu B, Ren J. Cardiac-specific overexpression of metallothionein rescues nicotine-induced cardiac contractile dysfunction and interstitial fibrosis. Toxicol Lett. 2011; 202: 8-14.[PMID: 21238558].
12. Rajiyah G, Agarwal R, Avendano G, Lyons M, Soni B, Regan TJ. Influence of nicotine on myocardial stiffness and fibrosis during chronic ethanol use. Alcohol Clin Exp Res. 1996; 20: 985-989. [PMID: 8892516].
13. Goette A, Lendeckel U, Kuchenbecker A, Bukowska A, Peters B, Klein HU, Huth C, Röcken C. Cigarette smoking induces atrial fibrosis in humans via nicotine. Heart. 2007; 93: 1056-1063. [PMC: 1955003].
14. Huang LT, Chou HC, Lin CM, Yeh TF, Chen CM, et al. Maternal Nicotine Exposure Exacerbates Neonatal Hyperoxia-Induced Lung Fibrosis in Rats. Neonatology. 2014; 106: 94-101. [PMID: 24851831].
15. Dasgupta C, Xiao D, Xu Z, Yang S, Zhang L. Developmental nicotine exposure results in programming of alveolar simplification and interstitial pulmonary fibrosis in adult male rats. Reprod Toxicol. 2012; 34: 370-377. [PMID: 22691361].
16. Jensen K, Afroze S, Ueno Y, Rahal K, Frenzel A, Sterling M, Guerrier M, et al. (2013) Chronic nicotine exposure stimulates biliary growth and fibrosis in normal rats. Dig Liver Dis. 45: 754-761. [PMID: 23587498].
17. Al-Wadei MH, Al-Wadei HA, Schuller HM. Pancreatic Cancer Cells and Normal Pancreatic Duct Epithelial Cells Express an Autocrine Catecholamine Loop that Is Activated by Nicotinic Acetylcholine Receptors alpha3, alpha5, and alpha7. Mol Cancer Res. 2012; 10: 239-49. [PMID: 22188668].
18. Browne CJ, Sharma N, Waters KA, Machaalani R. The effects of nicotine on the alpha-7 and beta-2 nicotinic acetycholine receptor subunits in the developing piglet brainstem. Int J Dev Neurosci. 2010; 28: 1-7. [PMID: 19896527].
19. Paulo JA, Brucker WJ, Hawrot E. Proteomic analysis of an alpha7 nicotinic acetylcholine receptor interactome. J Proteome Res. 2009; 8: 1849-1858. [PMID: 19714875].
20. Momi N, Ponnusamy MP, Kaur S, Rachagani S, Kunigal SS, Chellappan S, Ouellette MM, Batra SK. Nicotine/cigarette smoke promotes metastasis of pancreatic cancer through α7nAChR-mediated MUC4 upregulation. Oncogene. 2013; 32: 1384-1395. [PMID: 22614008].
21. Chu KM, Cho CH, Shin VY. Nicotine and gastrointestinal disorders: its role in ulceration and cancer development. Curr Pharm Des. 2013; 19: 5-10. [PMID: 22950507].
22. van Geenen EJ, Smits MM, Schreuder TC, van der Peet DL, Bloemena E, Mulder CJ. Smoking is related to pancreatic fibrosis in humans. Am J Gastroenterol. 2011; 106: 1161-1166. [PMID: 21577244].
23. Löhr M1, Trautmann B, Göttler M, Peters S, Zauner I, Maier A, Klöppel G, Liebe S, Kreuser ED. Expression and function of receptors for extracellular matrix proteins in human ductal adenocarcinomas of the pancreas. Pancreas. 1996; 12: 248-259. [PMID: 8830331].
24. Khoi PN, Park JS, Kim NH, Jung YD. Nicotine stimulates urokinase-type plasminogen activator receptor expression and cell invasiveness through mitogen-activated protein kinase and reactive oxygen species signaling in ECV304 endothelial cells. Toxicol Appl Pharmacol. 2012; 259: 248-256. [PMID: 22261521].
25. Wang Z, Wu W, Fang X, Wang Y, Xiao C, Zhao R, Wang L, Qiao Z. Protein expression changed by nicotine in rat vascular smooth muscle cells. J Physiol Biochem. 2007; 63: 161-169. [PMID: 17933390].
26. Lee KM, Nguyen C, Ulrich AB, Pour PM, Ouellette MM. Immortalization with telomerase of the Nestin-positive cells of the human pancreas. Biochem Biophys Res Commun. 2003; 301: 1038-1044. [PMID: 12589817].
27. Lieber M, Mazzetta J, Nelson-Rees W, Kaplan M, Todaro G. Establishment of a continuous tumor-cell line (panc-1) from a human carcinoma of the exocrine pancreas. Int J Cancer. 1975; 15: 741-747. [PMID: 1140870].
28. Paulo JA, Urrutia R, Banks PA, Conwell DL, Steen H. Proteomic analysis of a rat pancreatic stellate cell line using liquid chromatography tandem mass spectrometry (LC-MS/MS). J Proteomics. 2011; 75: 708-717. [PMID: 21968429].
29. Paulo JA, Urrutia R, Banks PA, Conwell DL, Steen H. Proteomic analysis of an immortalized mouse pancreatic stellate cell line identifies differentially-expressed proteins in activated vs nonproliferating cell states. J Proteome Res. 2011; 10: 4835-4844. [PMID: 21838295].
30. Ting L, Rad R, Gygi SP, Haas W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat Methods. 2011; 8: 937-940. [PMID: 21963607].
31. Elias JE, Gygi SP. Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol Biol. 2010; 604: 55-71. [PMID: 20013364].
32. Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villén J, Haas W, et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010; 143: 1174-1189. [PMID: 21183079].
33. Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B-Methodological. 1995; 57: 289-300.
34. Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003; 34: 374-378. [PMID: 12613259].
35. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009; 4: 44-57. [PMID: 19131956].
36. Mudbhary R, Hoshida Y, Chernyavskaya Y, Jacob V, Villanueva A, Fiel MI, Chen X, Kojima K. et al. UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer Cell. 2014; 25: 196-209. [PMID: 24486181].
37. Jensen K, Afroze S, Munshi MK, Guerrier M, Glaser SS. Mechanisms for nicotine in the development and progression of gastrointestinal cancers. Transl Gastrointest Cancer. 2012; 1: 81-87. [PMID: 22701817].
38. Dasgupta P, Rizwani W, Pillai S, Kinkade R, Kovacs M, Rastogi S, Banerjee S, Carless M, et al. Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int J Cancer. 2009; 124: 36-45. [PMID: 18844224].
39. Xu Y, Zhang Y, Cardell LO. Nicotine Exaggerates LPS-Induced Airway Hyperreactivity via JNK-Mediated Up-regulation of Toll-Like Receptor 4. Am J Respir Cell Mol Biol. 2014; 51: 370-379. [PMID: 24669857].
40. Jonnala RR, Terry AV Jr, Buccafusco JJ. Nicotine increases the expression of high affinity nerve growth factor receptors in both in vitro and in vivo. Life Sci. 2002; 70: 1543-1554. [PMID: 11895105].
41. Arany I1, Clark J, Reed DK, Juncos LA. Chronic nicotine exposure augments renal oxidative stress and injury through transcriptional activation of p66shc. Nephrol Dial Transplant. 2013; 28: 1417-1425. [PMID: 23328708].
42. Banerjee S, Chattopadhyay K, Chhabra JK, Chattopadhyay B. Protein dependent fate of hepatic cells under nicotine induced stress and curcumin ameliorated condition. Eur J Pharmacol. 2012; 684: 132-145. [PMID: 22381069].
43. Shimizu K. Mechanisms of pancreatic fibrosis and applications to the treatment of chronic pancreatitis. J Gastroenterol. 2008; 43: 823-832. [PMID: 19012035].
44. Apte MV, Wilson JS. Mechanisms of pancreatic fibrosis. Dig Dis. 2004: 22: 273-279. [PMID: 15753610].
45. Masamune A, Kikuta K, Watanabe T, Satoh K, Hirota M, Hamada S, Shimosegawa T. Fibrinogen induces cytokine and collagen production in pancreatic stellate cells. Gut. 2009; 58: 550-559. [PMID: 19052021].
46. Apte MV, Haber PS, Darby SJ, Rodgers SC, McCaughan GW, Korsten MA, Pirola RC, Wilson JS. Pancreatic stellate cells are activated by proinflammatory cytokines: implications for pancreatic fibrogenesis. Gut. 1999; 44: 534-541. [PMID: 10075961].
47. Kamer AR, El-Ghorab N, Marzec N, Margarone JE 3rd, Dziak R. Nicotine induced proliferation and cytokine release in osteoblastic cells. Int J Mol Med. 2006; 17: 121-127. [PMID: 16328020].
48. Hakki A, Hallquist N, Friedman H, Pross S. Differential impact of nicotine on cellular proliferation and cytokine production by LPS-stimulated murine splenocytes. Int J Immunopharmacol. 2000; 22: 403-410. [PMID: 10727751].
49. Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002; 416: 535-539. [PMID: 11932745].
50. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002; 297: 353-356. [PMID: 12130773].
51. Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD. Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal alpha7 nicotinic acetylcholine receptors: In vitro and in vivo mechanisms related to Alzheimer's disease. J Neurosci. 2001; 21: 4125-4133. [PMID: 11404397].
52. Wang HY, Lee DH, D'Andrea MR, Peterson PA, Shank RP, Reitz AB. beta-Amyloid(1-42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer's disease pathology. J Biol Chem. 2000; 275: 5626-5632. [PMID: 10681545].
53. Hoffmann J, Twiesselmann C, Kummer MP, Romagnoli P, Herzog V. A possible role for the Alzheimer amyloid precursor protein in the regulation of epidermal basal cell proliferation. Eur J Cell Biol. 2000; 79: 905-914. [PMID: 11152291].
54. Sandbrink R, Masters CL, Beyreuther K. Beta A4-amyloid protein precursor mRNA isoforms without exon 15 are ubiquitously expressed in rat tissues including brain, but not in neurons. J Biol Chem. 1994; 269: 1510-1517. [PMID: 8288617].
55. Ge YW, Lahiri DK. Regulation of promoter activity of the APP gene by cytokines and growth factors: implications in Alzheimer's disease. Ann N Y Acad Sci. 2002; 973: 463-467. [PMID: 12485912].
56. Paulo JA, Urrutia R, Kadiyala V, Banks P, Conwell DL, Steen H. Cross-species analysis of nicotine-induced proteomic alterations in pancreatic cells. Proteomics. 2013; 13: 1499-1512. [PMID: 23456891].
57. Hansel DE, Rahman A, Wehner S, Herzog V, Yeo CJ, Maitra A. Increased expression and processing of the Alzheimer amyloid precursor protein in pancreatic cancer may influence cellular proliferation. Cancer Res. 2003; 63: 7032-7037. [PMID: 14612490].
58. Barry PH, Lynch JW. Ligand-gated channels. IEEE Trans Nanobioscience. 2005; 4: 70-80. [PMID: 15816173].
59. Heeschen C, Weis M, Aicher A, Dimmeler S, Cooke JP. A novel angiogenic pathway mediated by non-neuronal nicotinic acetylcholine receptors. J Clin Invest. 2002; 110: 527-536. [PMID: 12189247].
60. Macklin KD, Maus AD, Pereira EF, Albuquerque EX, Conti-Fine BM. Human vascular endothelial cells express functional nicotinic acetylcholine receptors. J Pharmacol Exp Ther. 1998; 287: 435-439. [PMID: 9765366].
61. Sharma G, Vijayaraghavan S. Nicotinic receptor signaling in nonexcitable cells. J Neurobiol. 2002; 53: 524-534. [PMID: 12436417].
62. Arredondo J, Hall LL, Ndoye A, Nguyen VT, Chernyavsky AI, Bercovich D, Orr-Urtreger A, Beaudet AL. Central role of fibroblast alpha3 nicotinic acetylcholine receptor in mediating cutaneous effects of nicotine. Lab Invest. 2003; 83: 207-225. [PMID: 12594236].
63. Mai H, May WS, Gao F, Jin Z, Deng X. A functional role for nicotine in Bcl2 phosphorylation and suppression of apoptosis. J Biol Chem. 2003; 278: 1886-1891. [PMID: 12421819].
64. Chowdhury P, Udupa KB. Effect of nicotine on exocytotic pancreatic secretory response: role of calcium signaling. Tob Induc Dis. 2013; 11: 1. [PMID: 23327436].
65. Sevenich L, Schurigt U, Sachse K, Gajda M, Werner F, Müller S, Vasiljeva O, Schwinde A, et al. Synergistic antitumor effects of combined cathepsin B and cathepsin Z deficiencies on breast cancer progression and metastasis in mice. Proc Natl Acad Sci U S A. 2010; 107: 2497-2502. [PMID: 20133781].
66. Masamune A, Watanabe T, Kikuta K, Shimosegawa T. Roles of pancreatic stellate cells in pancreatic inflammation and fibrosis. Clin Gastroenterol Hepatol. 2009; 7: S48-54. [PMID: 19896099].
67. Kolodecik T, Shugrue C, Ashat M, Thrower EC. Risk factors for pancreatic cancer: underlying mechanisms and potential targets. Front Physiol. 2014; 4: 415. [PMID: 24474939].
68. Baker CH, Trevino JG, Summy JM, Zhang F, Caron A, Nesbit M, Gallick GE, Fidler IJ. Inhibition of PDGFR phosphorylation and Src and Akt activity by GN963 leads to therapy of human pancreatic cancer growing orthotopically in nude mice. Int J Oncol. 2006; 29: 125-138. [PMID: 16773192].
69. Shin VY, Wu WK, Chu KM, Koo MW, Wong HP, Lam EK, Tai EK, Cho CH. Functional role of beta-adrenergic receptors in the mitogenic action of nicotine on gastric cancer cells. Toxicol Sci. 2007; 96: 21-29. [PMID: 17003101].
70. Wei PL, Kuo LJ, Huang MT, Ting WC, Ho YS, Wang W, An J, Chang YJ. Nicotine enhances colon cancer cell migration by induction of fibronectin. Ann Surg Oncol. 2011; 18: 1782-1790. [PMID: 21210228].
71. Russell MA, Jarvis M, Iyer R, Feyerabend C. Relation of nicotine yield of cigarettes to blood nicotine concentrations in smokers. Br Med J. 1980; 280: 972-976. [PMID: 7417765].
72. Dasgupta P, Rizwani W, Pillai S, Kinkade R, Kovacs M, Rastogi S, Banerjee S, Carless M, et al. Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. International journal of cancer. Journal international du cancer. 2009; 124: 36-45.
73. Soeda J, Morgan M, McKee C, Mouralidarane A, Lin C, Roskams T, Oben JA. Nicotine induces fibrogenic changes in human liver via nicotinic acetylcholine receptors expressed on hepatic stellate cells. Biochemical and biophysical research communications. 2012; 417: 17-22.
74. Ebhardt HA. Selected reaction monitoring mass spectrometry: a methodology overview. Methods Mol Biol. 2014; 1072: 209-222. [PMID: 24136525].
75. Wasinger VC, Zeng M, Yau Y. Current status and advances in quantitative proteomic mass spectrometry. Int J Proteomics. 2013: 180605. [PMID: 23533757].